Сертификат GMP — это соблюдение изготовителем лекарственных препаратов требований надлежащей производственной практики. В России они сформулированы в национальном стандарте ГОСТ Р 52249-2009, который идентичен правилам, действующим в Европейском Союзе.
К каким производствам применима эта процедура?
В настоящее время в странах, которые контролируют соответствие стандарту GMP на своих территориях, его правила применяются для проверки качества следующих категорий продукции:
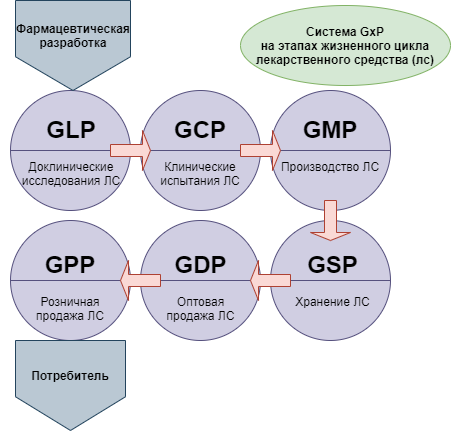
При этом для понимания ситуации следует принимать во внимание, что новая версия сертификации GMP — это не единственная система требований, которые в международной практике применяются в целях стандартизации медицинского обслуживания населения. Кроме них, производителям, работающим в такой сфере как фармация, необходимо соответствовать требованиям комплекса правил, объединенных под общим наименованием GxP:
Нормативная база
В Российской Федерации получение сертификата GMP осуществляется на основании действующей нормативной базы, включающей следующие основные правовые акты:
При этом необходимо принимать во внимание, что в настоящий момент наша страна вместе с другими государствами, входящими в состав Евразийского экономического союза, находится на этапе становления общего рынка, объединяющего фармацевтическое и косметическое производство в границах Союза. Это предполагает в том числе введение в действие единых требований к качеству и безопасности таких продуктов. В соответствии с принятым в мире порядком они реализуются в форме внедрения стандартов надлежащей производственной практики. Применение таких стандартов регулируется следующими нормативными документами:
Для полноценного применения разработанного административного регламента необходимо решение Правительства о порядке реализации некоторых процедур, связанных с проведением фармацевтических инспекций. Приказ № 2945 вступит в силу только после принятия соответствующего постановления: пока этого не произошло.
Преимущества обладания сертификатом
Несмотря на необходимость проведения достаточно сложной и дорогостоящей процедуры, производители знают, что сертификация по стандартам GMP является весьма важной для представителей фармацевтической отрасли. В частности, оно обеспечивает продукции и производству следующие серьезные преимущества:
Каков срок действия сертификата?
Срок действия российских сертификатов составляет 3 года. При этом срок действия иностранного сертификата GMP составляет от 1 до 3 лет. По истечении этого периода сертификацию потребуется проходить заново. Кроме того, это означает, что на протяжении всего этого срока компании необходимо обеспечить соответствие своего производства и продукции требованиям комплекса правил GMP.
Кто в России занимается сертификацией по стандартам GMP?
Сейчас сертификация контролируется департаментом развития медицинской и фармацевтической промышленности Министерства промышленности и торговли РФ. Он является организацией, ответственной за обеспечение надлежащего контроля за качеством, безопасностью и эффективностью лекарственных средств. Осуществлением требуемых сертификационных процедур занимается Государственный институт лекарственных средств и надлежащих практик (ФГБУ «ГИЛС и НП»).
Стандарт GMP в международной практике
Процесс сертификации на соответствие лекарственного препарата стандартам GMP в международной практике имеет комплексный характер, а ее основной целью является подтверждение безопасности и действенности продукции. В этой связи для достижения поставленной цели специалисты аккредитованных сертификационных организаций не ограничиваются оценкой ряда выборочных образцов лекарственных препаратов, как это часто предусматривается другими стандартами. В процедуру установления требуемого уровня качества лекарств любой международный центр сертификации лекарственных средств включает оценку предприятия, занимающегося его выпуском. В результате эксперты, занимающиеся проведением сертификации, анализируют конкретный препарат и процесс его выпуска в следующих областях:
Правила GMP в России
Порядок и сроки проведения всех операций в рамках этой процедуры, список лиц и организаций, ответственных за их осуществление, размер платы за проведение экспертной оценки и другие аспекты выполнения сертификации определены постановлением Правительства № 1314.
Процедура получения сертификата в России
Первым шагом для производителя, который желает пройти сертификацию, является подача соответствующего заявления в Минпромторг. В течение 10 рабочих дней специалисты ведомства проводят проверку корректности представленных в заявлении сведений и определяют возможность проведения сертификации.
В случае необходимости они вправе запросить у заявителя дополнительные документы, которые он обязан предоставить в течение 20 рабочих дней. В случае, если в отношении данного препарата принято положительное решение о проведении процедуры сертификации, необходимые данные направляются в ФГБУ «ГИЛС и НП», который в течение 20 рабочих дней с момента их получения обязан определить дату проведения сертификационных мероприятий и внести ее в график. Такая дата должна наступить не позднее 160 рабочих дней со дня, когда специалисты Минпромторга приняли положительное решение о сертификации, а сама экспертиза и расшифровка ее результатов должны занимать не более 10 рабочих дней.
На подготовку итогового отчета по результатам ее проведения исполнителю отводится 30 рабочих дней, а на его направление заявителю — 3 рабочих дня. Копия такого отчета также направляется в Минпромторг. На основании отчета формируется окончательное заключение, которое в случае положительного характера сопровождается выдачей сертификата производителю лекарственного препарата.
Документы для сертификации
Чтобы получить сертификат GMP в России, производитель обращается в уполномоченный орган с заявлением, к которому прилагает пакет документов, включающий:
Важнейшие документы предоставляются заявителем в копиях, поскольку при утере их восстановить невозможно или очень сложно. Правила регламентируют, что если заявление подает иностранный производитель, и некоторые документы в составе пакета представлены на другом языке, они должны быть переведены на русский язык и заверены в установленном порядке.
Сроки сертификации
Общая продолжительность процедуры сертификации складывается из следующих сроков.
160-дневный период инспектирования включает внесение производителя в график инспекций, ожидание процедуры и проведение самой инспекции. Она должна занимать не более 10 рабочих дней.
Такой порядок действует, если в документации, поданной производителем, не обнаружат ошибок и недочетов, из-за которых ее могут направить на доработку. В этом случае вся процедура займет немногим более 180 рабочих дней, то есть свыше 8 месяцев.
Стоимость получения сертификата
Обязательной для всех производителей лекарственных средств, претендующих на получение сертификата, подтверждающего соответствие их продукции стандартам GMP, является оплата государственной пошлины за рассмотрение соответствующего заявления в Министерстве промышленности и торговли. Ее размер составляет 7500 рублей. Оплатить данную сумму необходимо еще до подачи заявления в ведомство, а ее размер никак не зависит от результатов рассмотрения документа.
Однако данная пошлина — это далеко не единственный и не самый крупный платеж, который потребуется осуществить производителю лекарств. Другой значительной статьей расходов станет плата за проведение экспертной оценки производства и продукции заявителя. Такая процедура выполняется специалистами ФГБУ «ГИЛС и НП»: для каждого из них предварительно проводится аттестация эксперта по GMP в России.
При этом размер платы за проведение оценки не является строго установленным, а определяется в зависимости от объема, характера и сложности необходимых процедур в соответствии с положениями приказа Министерства промышленности и торговли Российской Федерации от 11.01.2016 № 9 «Об утверждении методики определения размера платы за оказание услуги по инспектированию GMP». В случае, если проверка потребует проведения значительного объема работы и привлечения большого количества высококвалифицированных экспертов, размер платы за ее проведение может превышать 2,5 миллиона рублей.
Лицензия на производство ветеринарных препаратов

Федеральная служба по ветеринарному и фитосанитарному надзору (Россельхознадзор)
Стоимость оформления лицензии на производство ветеринарных препаратов: от 20 000 рублей (в зависимости от пакета документов).Сроки оформления лицензии на производство ветеринарных препаратов: 45 рабочих дней + 5-10 дней на подготовку пакета документов.Государственная пошлина за получение лицензии:7500 рублей – Предоставление лицензии.3500 рублей — Переоформление лицензии.Территория действия лицензии: На всей территории Российской Федерации.Срок действия: Бессрочно.
В соответствии с Федеральным Законом РФ № 61-ФЗ от 12.04.2010 года «Об обращении лекарственных средств» (с изм. и доп., вступившими в силу с 01.07.2015 г.), а также Федеральным законом РФ № 429-ФЗ от 22.12.2014 года «О внесении изменений в Федеральный закон «Об обращении лекарственных средств» осуществление деятельности подлежит обязательному лицензированию в уполномоченных органах государственной власти.
ООО «ЦЕНТР ПРОФЕССИОНАЛОВ» оказывает помощь в получении, переоформлении лицензии на производство ветеринарных препаратов
1) Производство, хранение и реализация фармацевтических субстанций, получаемых методами химического синтеза. 2) Производство, хранение и реализация фармацевтических субстанций, получаемых методами биотехнологического синтеза. 3) Производство, хранение и реализация фармацевтических субстанций, получаемых методами выделения из химического сырья. 4) Производство, хранение и реализация фармацевтических субстанций, получаемых методами выделения из источников биологического, животного происхождения. 5) Производство, хранение и реализация фармацевтических субстанций, получаемых методами выделения из источников растительного происхождения. 6) Производство, хранение и реализация стерильных лекарственных препаратов с указанием конкретной лекарственной формы (аэрозоль, гель, крем, линимент, лиофилизированные продукты, мазь, паста, пленка, порошок, раствор, раствор для инъекций, стеклообразная масса, стерильная пористая масса, суспензия, сухая масса, таблетки, эмульсии). 7) Производство, хранение и реализация нестерильных лекарственных препаратов с указанием конкретной лекарственной формы (аэрозоль, бальзам, брикет, гель, гранулы, драже, капли, капсулы, капсулы мягкие, крем, линимент, мазь, масло, микрогранулы, микрокапсулы, настой, настойка, пастилки, паста, пеллеты, пластины, пластинки, пластырь, пленка, полимерная лента, полоски, порошок, раствор, сироп, спрей, суппозитории, суспензия, таблетки, шнур, экстракт, эликсир, эмульсия).
Необходимые документы. Лицензия на производство ветеринарных препаратов
Заявление, в котором указывается:
1) полное и (в случае, если имеется) сокращенное наименование, в том числе фирменное наименование, и организационно-правовая форма юридического лица, адрес его места нахождения, адреса мест осуществления лицензируемого вида деятельности, который намерен осуществлять соискатель лицензии, государственный регистрационный номер записи о создании юридического лица, данные документа, подтверждающего факт внесения сведений о юридическом лице в единый государственный реестр юридических лиц, с указанием адреса места нахождения органа, осуществившего государственную регистрацию, а также номера телефона и (в случае, если имеется) адреса электронной почты юридического лица;
2) идентификационный номер налогоплательщика и данные документа о постановке соискателя лицензии на учет в налоговом органе;
3) лицензируемый вид деятельности, который соискатель лицензии намерен осуществлять;
4) реквизиты документа, подтверждающего уплату государственной пошлины за рассмотрение лицензирующим органом заявления о предоставлении лицензии;
5) копии документов, подтверждающих наличие у соискателя лицензии на праве собственности или на ином законном основании необходимых для осуществления деятельности по производству лекарственных средств помещений, зданий, сооружений и иных объектов, технических средств, оборудования и технической документации, соответствующих установленным требованиям, права на которые не зарегистрированы в Едином государственном реестре прав на недвижимое имущество и сделок с ним;
6) копии титульных листов промышленных регламентов;
7) копии документов, подтверждающих соответствующие лицензионным требованиям образование, квалификацию и стаж работы уполномоченного лица производителя лекарственных средств, а также образование специалистов, ответственных за производство и маркировку лекарственных средств;
8) опись прилагаемых документов.
В случае изменения наименования юридического лица или места его нахождения, в заявлении о переоформлении лицензии указываются новые сведения о лицензиате и данные документа, подтверждающего факт внесения соответствующих изменений в единый государственный реестр юридических лиц (для лицензиата — юридического лица), в единый государственный реестр индивидуальных предпринимателей.
1) заявление о переоформлении лицензии;
2) оригинал действующей лицензии;
3) копии документов, подтверждающих повышение квалификации (при наличии новых специалистов копии документов о высшем или среднем фармацевтическом или ветеринарном образовании, о стаже работы по соответствующей специальности и сертификата специалиста).
Помимо указанных документов направляются следующие документы в соответствии с причиной переоформления:
При намерении осуществлять новые работы, составляющие деятельность по производству лекарственных средств, ранее не указанных в лицензии,
а) сведения о составляющих деятельность по производству лекарственных средств новых работах, которые лицензиат намерен выполнять;
б) сведения о наличии у лицензиата на праве собственности или на ином законном основании необходимых для осуществления новых работ, составляющих деятельность по производству лекарственных средств, помещений, зданий, сооружений и иных объектов, технических средств, оборудования и технической документации, соответствующих установленным требованиям, а также промышленных регламентов;
в) сведения о наличии работников, заключивших трудовые договоры, имеющих соответствующее высшее или среднее профессиональное образование, ответственных за производство и маркировку лекарственных средств, намеренных осуществлять новые работы, составляющие деятельность по производству лекарственных средств.
При намерении осуществлять деятельность по производству лекарственных средств по адресу, не указанному в лицензии:
а) сведения, содержащие новый адрес осуществления деятельности по производству лекарственных средств;
б) сведения о составляющих деятельность по производству лекарственных средств работах, которые лицензиат намерен выполнять по новому адресу осуществления деятельности по производству лекарственных средств;
в) сведения о наличии у лицензиата на праве собственности или на ином законном основании необходимых для осуществления деятельности по производству лекарственных средств по указанному новому адресу помещений, зданий, сооружений и иных объектов, технических средств, оборудования и технической документации, соответствующих установленным требованиям, а также промышленных регламентов;
г) сведения о наличии работников, заключивших трудовые договоры, имеющих соответствующее высшее или среднее профессиональное образование, ответственных за производство и маркировку лекарственных средств, намеренных осуществлять деятельность по указанному новому адресу.
*Данный перечень не является исчерпывающим и уточняется в каждом конкретном случае.
Требования к лицензиату. Лицензия на производство ветеринарных препаратов
а) наличие у соискателя лицензии помещений, зданий, сооружений и иных объектов, технических средств, оборудования и технической документации, принадлежащих ему на праве собственности или на ином законном основании, необходимых для выполнения заявляемых работ, соответствующих установленным требованиям;
б) соответствие производства лекарственных средств правилам организации производства и контроля качества лекарственных средств в соответствии со статьей 45 Федерального закона «Об обращении лекарственных средств»;
в) наличие в соответствии со статьей 45 Федерального закона «Об обращении лекарственных средств» промышленных регламентов, утвержденных руководителем производителя лекарственных средств (соискателя лицензии) и включающих в себя перечень используемых фармацевтических субстанций и вспомогательных веществ с указанием количества каждого из них, данные об используемом оборудовании и описание технологического процесса и методов контроля на всех этапах производства лекарственных средств;
г) наличие в соответствии со статьей 45 Федерального закона «Об обращении лекарственных средств» уполномоченного лица производителя лекарственных средств, которое при вводе лекарственных средств в гражданский оборот осуществляет подтверждение соответствия лекарственных средств требованиям, установленным при их государственной регистрации, и гарантирует, что лекарственные средства произведены в соответствии с правилами производства и контроля качества лекарственных средств, а также которое:
имеет высшее фармацевтическое, химическое, медицинское или биологическое образование либо при производстве лекарственных средств для ветеринарного применения — ветеринарное образование, стаж работы не менее чем 5 лет в области производства и контроля качества лекарственных средств;
при производстве лекарственных средств для медицинского применения аттестовано в порядке, установленном Министерством здравоохранения Российской Федерации, при производстве лекарственных средств для ветеринарного применения — в порядке, установленном Министерством сельского хозяйства Российской Федерации;
д) наличие работников, заключивших трудовые договоры, имеющих соответственно высшее или среднее профессиональное фармацевтическое, химическое, химико-технологическое, биологическое, биотехнологическое, медицинское или ветеринарное образование, ответственных за производство и маркировку лекарственных средств.
Примеры форм заявлений. Лицензия на производство ветеринарных препаратов
Реестр лицензий Россельхознадзора
Преимущества работы с нами
Помощь на всех этапах формирования пакета документов для подачи в лицензионный орган (в том числе в оформлении недостающих документов, подготовке и обучении специалистов)
Мы бесплатно проконсультируем Вас по всем вопросам, касающихся получения лицензии
Несколько вариантов оплаты, в том числе поэтапная оплата работ
Надежность и ответственность
Мы заключаем юридически грамотно подготовленный договор, в котором прописаны виды работы, порядок оплаты и наши обязательства по его исполнению
Сопровождаем на всех этапах процедуры оформления лицензии, взаимодействуем с лицензионным органом
Отзывы клиентов о нашей работе
1. Производство лекарственных средств должно соответствовать требованиям правил надлежащей производственной практики, утвержденным уполномоченным федеральным органом исполнительной власти. Особенности перехода производства отдельных лекарственных средств к их производству в соответствии с требованиями правил надлежащей производственной практики устанавливаются Правительством Российской Федерации. Выдача заключений о соответствии производителя лекарственных средств требованиям правил надлежащей производственной практики осуществляется по результатам инспектирования производителей лекарственных средств в порядке, установленном Правительством Российской Федерации. Размер платы за выдачу заключения о соответствии производителя лекарственных средств требованиям правил надлежащей производственной практики устанавливается Правительством Российской Федерации. Порядок организации и проведения инспектирования производителей лекарственных средств на соответствие требованиям правил надлежащей производственной практики устанавливается Правительством Российской Федерации.
(в ред. Федеральных законов от 25.06.2012 N 93-ФЗ, от 22.10.2014 N 313-ФЗ, от 22.12.2014 N 429-ФЗ)
(см. текст в предыдущей редакции)
2. Производство лекарственных средств в Российской Федерации осуществляется производителями лекарственных средств, имеющими лицензию на производство лекарственных средств. Подтверждение соответствия лицензиата правилам надлежащей производственной практики осуществляется в рамках лицензионного контроля в соответствии с законодательством Российской Федерации с учетом особенностей, указанных в части 1 настоящей статьи.
(в ред. Федеральных законов от 22.10.2014 N 313-ФЗ, от 22.12.2014 N 429-ФЗ)
2.1. Для целей лицензирования производства лекарственных средств для медицинского применения и инспектирования субъектов обращения лекарственных средств на соответствие требованиям правил надлежащей производственной практики федеральный орган исполнительной власти, осуществляющий государственную регистрацию лекарственных препаратов для медицинского применения, предоставляет федеральному органу исполнительной власти, осуществляющему федеральный государственный лицензионный контроль деятельности по производству лекарственных средств для медицинского применения, по его запросу в рамках межведомственного информационного взаимодействия в порядке, установленном федеральным органом исполнительной власти, осуществляющим лицензирование производства лекарственных средств для медицинского применения, и федеральным органом исполнительной власти, осуществляющим государственную регистрацию лекарственных препаратов для медицинского применения, сведения о лекарственных средствах, указанные в части 5 статьи 18 настоящего Федерального закона, в том числе в случае, если такие сведения составляют коммерческую тайну.
(часть 2.1 введена Федеральным законом от 14.07.2022 N 311-ФЗ)
3. Производство лекарственных средств осуществляется с соблюдением требований промышленного регламента, который утверждается руководителем производителя лекарственных средств и включает в себя перечень используемых фармацевтических субстанций и вспомогательных веществ с указанием количества каждого из них, данные об используемом оборудовании, описание технологического процесса и методов контроля на всех этапах производства лекарственных средств.
4. При производстве лекарственных средств используются фармацевтические субстанции, сведения о которых содержатся в государственном реестре лекарственных средств, за исключением фармацевтических субстанций, производимых для проведения клинических исследований и для экспорта. К процессу производства фармацевтической субстанции относятся любые стадии технологического процесса, позволяющие получить готовый продукт, соответствующий требованиям фармакопейной статьи, в том числе ферментация, экстракция, очистка, выделение, перекристаллизация, высушивание, измельчение. В случае необходимости использования этилового спирта, в том числе фармацевтической субстанции спирта этилового (этанола), при производстве лекарственных средств в качестве действующего и (или) вспомогательного вещества, а также в иных технологических целях должна быть использована только фармацевтическая субстанция спирта этилового (этанол).
(в ред. Федеральных законов от 22.12.2014 N 429-ФЗ, от 27.12.2019 N 462-ФЗ, от 27.12.2019 N 481-ФЗ)
4.1. Перечень лекарственных препаратов для медицинского применения, в отношении которых устанавливаются требования к объему тары, упаковке и комплектности, перечень лекарственных препаратов для ветеринарного применения, в отношении которых устанавливаются требования к объему тары, а также требования к объему тары, упаковке и комплектности лекарственных препаратов для медицинского применения, требования к объему тары лекарственных препаратов для ветеринарного применения определяются соответствующими уполномоченными федеральными органами исполнительной власти в порядке, установленном Правительством Российской Федерации.
(часть 4.1 в ред. Федерального закона от 14.12.2015 N 374-ФЗ)
4.2. Организации, осуществляющие производство фармацевтической субстанции спирта этилового (этанола) для производства спиртосодержащих лекарственных препаратов, обязаны осуществлять учет в порядке, установленном статьями 8 и 14 Федерального закона от 22 ноября 1995 года N 171-ФЗ “О государственном регулировании производства и оборота этилового спирта, алкогольной и спиртосодержащей продукции и об ограничении потребления (распития) алкогольной продукции”. Производство фармацевтической субстанции спирта этилового (этанола) допускается только организацией, осуществляющей производство ректификованного этилового спирта из пищевого сырья на основании лицензии на производство этилового спирта для производства фармацевтической субстанции спирта этилового (этанола) в соответствии с требованиями Федерального закона от 22 ноября 1995 года N 171-ФЗ “О государственном регулировании производства и оборота этилового спирта, алкогольной и спиртосодержащей продукции и об ограничении потребления (распития) алкогольной продукции”, методом разведения водой очищенной такого этилового спирта, произведенного данной организацией по месту осуществления производства фармацевтической субстанции спирта этилового (этанола).
(часть 4.2 введена Федеральным законом от 03.07.2016 N 261-ФЗ; в ред. Федерального закона от 27.12.2019 N 481-ФЗ)
4.3. Индивидуальные предприниматели и юридические лица, осуществляющие закупку и использование фармацевтической субстанции спирта этилового (этанола), а также производство, изготовление и (или) оборот (за исключением розничной продажи) спиртосодержащих лекарственных препаратов, обязаны осуществлять их учет и декларирование в порядке, установленном статьями 8 и 14 Федерального закона от 22 ноября 1995 года N 171-ФЗ “О государственном регулировании производства и оборота этилового спирта, алкогольной и спиртосодержащей продукции и об ограничении потребления (распития) алкогольной продукции”.
(часть 4.3 введена Федеральным законом от 29.07.2017 N 278-ФЗ)
4.4. При производстве лекарственных препаратов для ветеринарного применения допускается использование произведенных для реализации фармацевтических субстанций, включенных в государственный реестр лекарственных средств уполномоченным федеральным органом исполнительной власти, осуществляющим ведение государственного реестра лекарственных средств для медицинского применения.
(часть 4.4 введена Федеральным законом от 02.08.2019 N 297-ФЗ)
5. Запрещается производство:
1) лекарственных средств, не включенных в государственный реестр лекарственных средств, за исключением лекарственных средств, производимых для проведения клинических исследований и для экспорта;
2) фальсифицированных лекарственных средств;
3) лекарственных средств без лицензии на производство лекарственных средств;
4) лекарственных средств с нарушением правил организации производства и контроля качества лекарственных средств;
5) утратил силу. – Федеральный закон от 27.12.2019 N 481-ФЗ;
6) спиртосодержащих лекарственных препаратов по месту осуществления производства фармацевтической субстанции спирта этилового (этанола) и (или) по месту осуществления производства этилового спирта;
(п. 6 введен Федеральным законом от 27.12.2019 N 481-ФЗ)
Федеральным законом от 22.12.2020 N 436-ФЗ ст. 14.1 ФЗ от 22.11.1995 N 171-ФЗ изложена в новой редакции, норма п. 1.1 ст. 14.1 старой редакции ФЗ соответствует норме п. 2 ст. 14.1 новой редакции.
7) фармацевтической субстанции спирта этилового (этанола) и (или) спиртосодержащих лекарственных препаратов на основном технологическом оборудовании для производства этилового спирта, указанном в пункте 1.1 статьи 14.1 Федерального закона от 22 ноября 1995 года N 171-ФЗ “О государственном регулировании производства и оборота этилового спирта, алкогольной и спиртосодержащей продукции и об ограничении потребления (распития) алкогольной продукции”.
(п. 7 введен Федеральным законом от 27.12.2019 N 481-ФЗ)
6. При вводе лекарственных средств в гражданский оборот уполномоченное лицо производителя лекарственных средств осуществляет подтверждение соответствия лекарственных средств требованиям, установленным при их государственной регистрации, и гарантирует, что лекарственные средства произведены в соответствии с требованиями правил надлежащей производственной практики.
(в ред. Федерального закона от 22.12.2014 N 429-ФЗ)
7. Уполномоченным лицом производителя лекарственных средств является его работник, аттестованный в установленном уполномоченным федеральным органом исполнительной власти порядке и имеющий стаж работы не менее чем пять лет в области производства и (или) контроля качества лекарственных средств, высшее образование соответственно по одной из специальностей и (или) одному из направлений подготовки: биология, биотехнология, ветеринария, ветеринарно-санитарная экспертиза, клиническая медицина, радиационная, химическая и биологическая защита, фармация, фундаментальная медицина, химическая технология, химия.
(в ред. Федеральных законов от 22.10.2014 N 313-ФЗ, от 02.08.2019 N 297-ФЗ)
8. Производители лекарственных средств (за исключением фармацевтической субстанции спирта этилового (этанола) могут осуществлять продажу лекарственных средств или передавать их в установленном законодательством Российской Федерации порядке:
(в ред. Федерального закона от 29.07.2017 N 278-ФЗ)
1) другим производителям лекарственных средств для производства лекарственных средств;
2) организациям оптовой торговли лекарственными средствами;
3) аптечным организациям, ветеринарным аптечным организациям, индивидуальным предпринимателям, имеющим лицензию на фармацевтическую деятельность или лицензию на медицинскую деятельность;
4) научно-исследовательским организациям для научно-исследовательской работы;
5) медицинским организациям и ветеринарным организациям;
6) организациям и индивидуальным предпринимателям, осуществляющим разведение, выращивание и содержание животных (за исключением продажи им фармацевтических субстанций или их передачи).
(п. 6 в ред. Федерального закона от 02.08.2019 N 297-ФЗ)
9. Производители фармацевтической субстанции спирта этилового (этанола) обязаны осуществлять реализацию (передачу в установленном законодательством Российской Федерации порядке) фармацевтической субстанции спирта этилового (этанола) производителям лекарственных средств для производства лекарственных средств, производителям (изготовителям) медицинских изделий для производства и изготовления медицинских изделий в емкостях объемом не более 1 литра и (или) не менее 1000 литров, а организациям, указанным в пунктах 3 – 5 части 8 настоящей статьи, в таре объемом не более 1 литра.
(часть 9 введена Федеральным законом от 29.07.2017 N 278-ФЗ; в ред. Федеральных законов от 27.12.2019 N 481-ФЗ, от 22.12.2020 N 444-ФЗ)
10. Реализация (передача в установленном законодательством Российской Федерации порядке) фармацевтической субстанции спирта этилового (этанола) организациям оптовой торговли лекарственными средствами не допускается.
(часть 10 введена Федеральным законом от 27.12.2019 N 481-ФЗ)
11. Перевозки фармацевтической субстанции спирта этилового (этанола) осуществляются с соблюдением требований, установленных абзацем четвертым пункта 3 статьи 9 Федерального закона от 22 ноября 1995 года N 171-ФЗ “О государственном регулировании производства и оборота этилового спирта, алкогольной и спиртосодержащей продукции и об ограничении потребления (распития) алкогольной продукции”.
(часть 11 введена Федеральным законом от 27.12.2019 N 481-ФЗ)
I. Все производственные
процессы должны быть четко регламентированы
и должны периодически пересматриваться
с учетом накопленного опыта. Следует
контролировать стабильность производства
лекарственных средств с заданным
качеством в соответствии со спецификациями
на них.
– обученного и
аттестованного персонала; – необходимых
помещений и площадей;
– соответствующего
оборудования и системы обслуживания;
– материалов,
средств упаковки и маркировки,
удовлетворяющих установленным
требованиям;
– утвержденных
инструкций и методик;
– требуемых условий
хранения и транспортирования.
IV. Инструкции и
методики должны быть конкретными,
изложены ясно и однознач-но в письменной
форме.
V. Персонал должен
быть обучен правильному выполнению
инструкций.
VI. В процессе
производства следует составлять
протоколы (заполняемые рукопис-ным
способом и/или с применением технических
средств), документально подтверждаю-щие
фактическое проведение предусмотренных
инструкциями технологических стадий
и получение продукции требуемого
качества в количестве, соответствующем
установленным нормам. Все отклонения
необходимо расследовать и протоколировать
в полном объеме.
VII. Протоколы на
серию продукции, в т.ч. на документацию
по реализации продук-ции, должны давать
возможность прослеживать изготовление
каждой серии и храниться в полном объеме
в доступной форме.
VIII. Порядок
реализации (оптовой продажи) продукции
должен сводить к миниму-му любой риск
для ее качества.
IX. Следует
организовать систему отзыва любой серии
продукции из продажи или поставки.
Приведите различия и общие принципы правил gmp, gcp и glp.
Надлежащая
лабораторная практика (GLP)

